
pyLFDA is a tool which allows analysis of pairwise lipid force distribution along with other functions such as curvature and diffusion. Our tools enables easy usage of different versions of Gromacs-FDA as the user is only required to specify the version and pyLFDA handles the rest. We provide 3 modes of usage for all properties -
- Average - Averages the property for each atom over all input frames.
- Framewise - Property values for a particular frame.
- Moving Window - Averages the property for each atom over a window of frames of specified size for all non overlapping windows possible.
pyLFDA has 3 different interfaces - GUI, CLI and Python package, each providing the same functionality while suiting your use case. We utilize force calculation done by Gromacs FDA and parse it into useful formats. pyLFDA also enable users to select the version of Gromacs FDA they require with one simple argument and it handles the rest for all future experiments.
This project works with Python on versions 3.5+, and on Linux, OSX and the Windows operating systems. To install:
pip install pyLFDA
Other requirements -
pyLFDA provides plots of pairwise force on selected residue groups. Forces calculated are plotted along the z - axis which helps users understand how forces are distributed along the membrane.
pyLFDA utilizes MDAnalysis and provides cross sectional plots along the x-y axis for the z-surface, curvature, gaussian curvature of the P atoms. We provide line plots for the bins containing the maximum and minimum values. Also, cross sectional force plots are provided which allows the user to find the correlation between forces on atoms and their curvature. Additional options include splitting the membranes into upper and lower membranes and changing the number of bins to divide the membrane cross section into.
pyFDA allows user to create PDB files with bFactor calculated either for individual atoms or averaged for each selected residue group.
We use MDAnalysis to calculate MSD and diffusion over all the frames.
For each experiment run, the plots and files are saved in a folder specified by the -experiment_name argument. If not specified, the program generates a folder for the time of program run. If a new Gromacs FDA version is supplied, it is downloaded into a new directory and it utilized for all future experiments. Gromacs FDA generates a .pfa file as output which generally is time consuming and for this we provide an argument -pfa which can load this file and does not require FDA to run again. pyLFDA further parses this file into averaged and framewise version both of which can be loaded using -avg_pfa and -f_pfa arguments respectively if one needs to do additional experiments on the same simulation.
Command Line Interface for pyLFDA. Download pyLFDA_cli.py(download) from the releases and follow the instructions from below.
usage: pyLFDA_cli.py [-h] -v Version [-exp Experiment Name] -trr TRR Filename
-tpr TPR Filename -ndx NDX Filename -pdb PDB Filename
-gro GRO Filename [-pfa PFA Filename]
[-avg_pfa Average Parsed PFA Filename]
[-f_pfa Frameise Parsed PFA Filename] [-avg]
[-f Specific Frame] [-window Moving Window] -gr1 Group 1
-gr2 Group 2 [-force] [-curve] [-diffu] [-cluster]
[-split] [-bfac bFactor] [-xbins Num_xBins]
[-ybins Num_yBins]
Command Line Interface for pyLFDA
required arguments:
-v Version Release version of Gromacs FDA to be used
-trr TRR Filename TRR file to be used
-tpr TPR Filename TPR file to be used
-ndx NDX Filename NDX file to be used
-pdb PDB Filename PDB file to be used
-gro GRO Filename GRO file to be used
optional arguments:
-h, --help show this help message and exit
-exp Experiment Name Name of the experiment. If not specified time-stamp of
experiment will be used
-pfa PFA Filename PFA file to be used. If PFA file is specified, FDA
wont run again
-avg_pfa Average Parsed PFA Filename
Average Parsed PFA file to be used. If Average PFA
file is specified, FDA and PFA parsing wont run again
-f_pfa Frameise Parsed PFA Filename
Frameise Parsed PFA file to be used. If PFA file is
specified, FDA and PFA parsing FDA wont run again
-avg Calculate average forces for all frames
-f Specific Frame Calculate forces for a specific frame
-window Moving Window
Calculate forces for a moving window
-gr1 Group 1 Group 1 to be selected
-gr2 Group 2 Group 2 to be selected
-force Calculate Force
-curve Calculate Curvature
-diffu Calculate Diffusion
-cluster Generate Lipid Cluster Plots
-angle ANGLE [ANGLE ...]
Calculate angle of selected lipids with the z-axis
-c_atom C_ATOM Name of the atom to which the lipid vector is to be defined
-split Split Calculations into Upper and Lower Membranes
-combine Combine lipids specifed for angle calculation
-bfac bFactor Calculate B-factor. "atomwise", "groupwise".
-xbins Num_xBins Number of bins in x-direction
-ybins Num_yBins Number of bins in y-direction
The GUI offers the same functionality with the ease of a graphical interface. To download, either clone the repository and run pyLFDA.exe or click here.
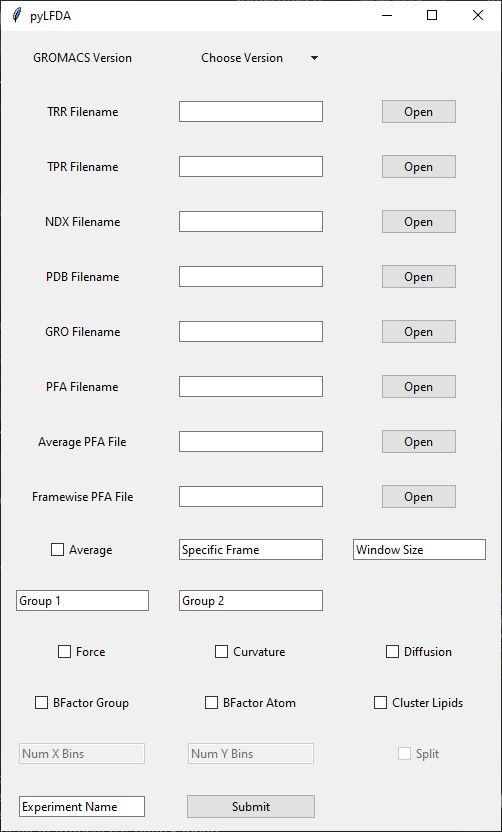
Detailed documentation and usage instructions for pyLFDA can be found in example-1, example-2, example-3.