PatternSearchingModel
This document assumes that you already know how to construct a pattern, but don't know how to search BioPAX models using the pattern.
If you are already familiar with using Paxtools, then below part-code should demonstrate searching the pattern in a model.
Pattern p = new Pattern(... // construct the pattern
Model model = ... // Get the model
// Search the pattern in the model.
Map<BioPAXElement, List<Match>> map = Searcher.search(model, p);
Match m = ... // next Match
BioPAXElement element = m.get("label of the element", p);
The Match object contains a matching result of the pattern search. Assigned values (BioPAX elements) can be accessed using the same labels used during constructing the pattern.
We developed the package org.biopax.paxtools.pattern.miner both as an example use of this framework, and as an easy entry point for new users who just want to define and search a pattern in a model quickly.
Assume we want to detect all protein interactions in a BioPAX model. This is not a straightforward task because simply iterating complexes and processing their members is not sufficient. Complex memberships can be arbitrarily nested, and membership of a protein can be through a generic entity. Below is the minimal code that can be used for that purpose.
The Miner object is capable of constructing the pattern and writing text result from the resulting Match objects. The Dialog class then handles the rest through a GUI.
// Define and initialize the miner.
Miner miner = new MinerAdapter("Appear-in-same-complex", "The pattern captures two " +
"proteins appear to be members of the same complex. There may be a nesting hierarchy " +
"in the complex, and the proteins can be represented with generic entities, again " +
"through multiple generic-member relations.")
{
/**
* The pattern is composed of two proteins associated to a complex as members. The
* relation can be through nested memberships or through generic relations.
*/
public Pattern constructPattern()
{
Pattern p = new Pattern(ProteinReference.class, "Protein 1");
p.add(ConBox.erToPE(), "Protein 1", "SPE1");
p.add(ConBox.linkToComplex(), "SPE1", "Complex");
p.add(new Type(Complex.class), "Complex");
p.add(ConBox.linkToSimple(), "Complex", "SPE2");
p.add(ConBox.peToER(), "SPE2", "Protein 2");
p.add(ConBox.equal(false), "Protein 1", "Protein 2");
p.add(new Type(ProteinReference.class), "Protein 2");
return p;
}
/**
* Writes the result as "P1 P2", where P1 and P2 are gene symbols of proteins and the
* whitespace is tab. The relation is undirected, so "P2 P1" is treated as the same
* relation.
*/
public void writeResult(Map<BioPAXElement, List<Match>> matches, OutputStream out)
throws IOException
{
writeResultAsSIF(matches, out, false, "Protein 1", "Protein 2");
}
};
// Launch the GUI that will assist choosing a source model, and output file name
Dialog d = new Dialog(miner);
d.setVisible(true);
This code is also available in test sources as a class named org.biopax.paxtools.pattern.miner.MinerTest.
The GUI will appear, where the user can select a model file, and output filename. This dialog can also automatically download and use Pathway Commons data. If the source model is too big then consider running the code using the virtual machine parameter -Xmx and increase the maximum heap size as necessary. Loading the complete Pathway Commons data will require more than 4GB memory.
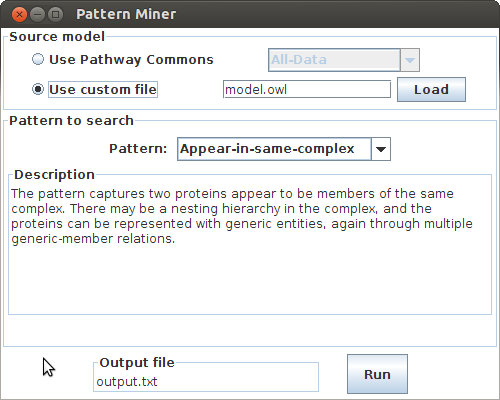
Feel free to modify this code with your own pattern and your way of handling the result.