PMID:27856494 transcriptome data (Kuang,Boeke,Canzar) #2296
Comments
https://pubmed.ncbi.nlm.nih.gov/27856494/ |
|
fixed PMID in header. I had used the example PMID. The template probably doesn't need examples of PMIDs! |
|
@ValWood Not entirely sure of what feedback you expect here, but I made a version of the template where I have filled the informations I could find. |
|
Sorry I didn't explain this well. Can discuss on the first call int eh New Year |
|
Li-Lin Du has sent us the mapped BAM files for this paper. (At least I think that's what he sent to us) I've processed the BAM files a bit to change the chromosome IDs in the files match what JBrowse needs. I changed "MTR" to "mating_type_region" and "MT" to "mitochondrial". |
|
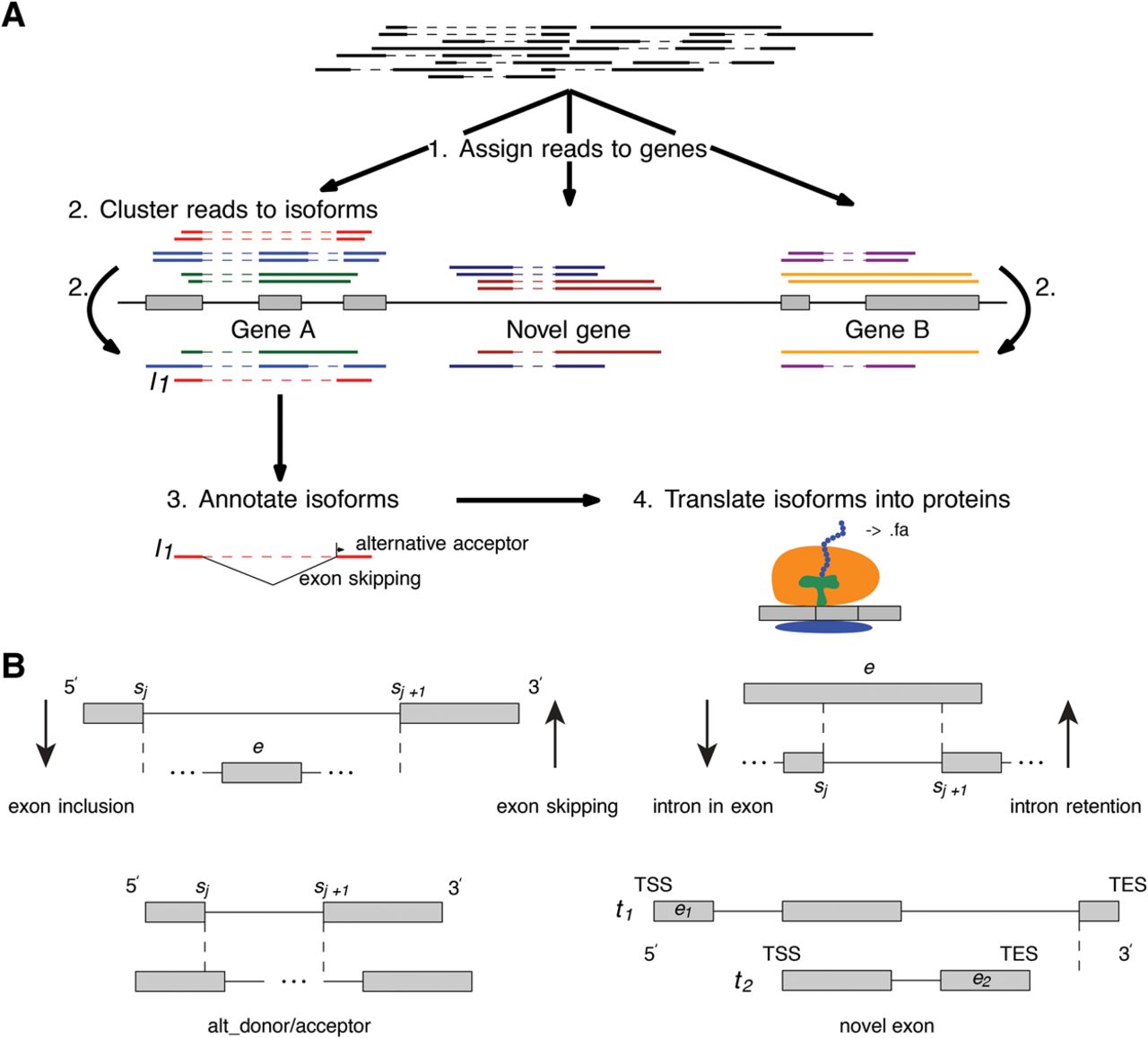
The BAM files from Li-Lin Du's group are raw mapped PacBio reads from transcripts. I don't think we have a dataset like that in JBrowse. The screenshot below is from a short read dataset. The PacBio reads will be much more joined-up, often spanning the whole gene. It will interesting to see how that looks in JBrowse. The paper is partly about their new tool ("SpliceHunter"). It sounds like it clusters the reads into possible isoforms and annotates the isoforms exon and intron changes like exon skipping and intron retention. I'm getting that from this diagram: The dataset in GEO has this supplementary file with details of the possible isoforms: GSE79802_isoforms.txt.gz I haven't worked out what all the columns mean yet. :-) We might want to consider turning that isoforms file into a GFF3 file (or several) which would summarise the reads and be faster to load.
|

Maybe not. I've just re-read the email from Li-Lin (subject: "The dynamic landscape of fission yeast meiosis alternative-splice isoforms"). He think the summary might lose information:
In the email he also has a screenshot of the reads loaded into JBrowse so we can get some idea about how it would look. The screenshot also has a coverage track which would good to show an overview of read depth. Note to self, there is a guide for that at the end of the docs page: https://github.com/pombase/website/wiki/Formatting-data-files-for-JBrowse#generating-bigwig-coverage-graphs-for-use-at-lower-zoom-levels I was looking in his email to understand why the GEO record has 18 samples (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79802), but there are only 6 BAM files. Perhaps there are replicates that have been combined. I should read the paper. :-) |
I think it would make for a clearer track, although as Li-Lin mentioned, we would lose the abundance measure. I guess there wouldn't really be a way to combine both on the same track ? |
|
I've made a start on showing the reads in JBrowse. Pascal, could you have a look at the metadata file when you have time to see if you can improve it?: The black lines are cases where a read has been mapped to the sequence with a large gap. I think we might want to filter those.
|

{kind=link}
https://pubmed.ncbi.nlm.nih.gov/27856494/
Format for track label “Assayed gene product” “Data type” “in mutant” “during Growth phase or response” “additional experimental detail of importance (Conditions, Strain background)” “; repeat” “(strand)” “- First author (Publication year)”
URL for dataset
Other info
transferred from #1441 (comment)
Li-Lin has lab has mapped the reads of this dataset to the reference genome. We will be happy to provide the mapping results to PomBase for loading into the genome browser.
Some novel genes are reported (but I think we likely have them all from Dannys data IIRC)
The text was updated successfully, but these errors were encountered: