RietveldRefinement
Rietveld refinement is a technique for minimising the differences between the modelled crystal data and the experimental powder pattern. This means that it is a very useful method for validating DASH solutions and for producing publication-quality crystal structures.
There are currently a large number of different programs available for performing Rietveld refinement. Within DASH there is a built-in rigid-body refinement module and interfaces to three external programs; TOPAS, GSAS and RIETAN. These interfaces are designed to facilitate the transfer of structural data to the external program and allow an inexperienced user to carry out a basic structural refinement. Each of the interfaces will not necessarily provide the best route through a refinement for any specific structure but are intended to offer a starting point for using the particular program.
No support is provided for the external programs, which are supported to differing extents by their own developers. The interfaces from DASH to each program are also not officially supported. The built-in rigid-body refinement module will however be supported and maitained.
Web-links for external programs:
There are currently four available options for Rietveld refinement in DASH.
Firstly, there is a built-in module for performing rigid-body Rietveld refinement which uses the rigid bodies (Z-matrices) from the simulated annealing stage. The use of rigid bodies imposes a large number of constraints on the atomic coordinates, which makes it more likely that the Rietveld refinement will result in a chemically reasonable crystal structure. Another advantage of this approach is that it is not necessary to include the atomic resolution data, so that the background and peak shape parameters from the Pawley refinement can be used.
Rietveld refinement can also be carried out in a semi-automated way using the DASH interface to one of three external programs; TOPAS, GSAS and RIETAN. These three interfaces prepare the reflection data for the external programs, set up the refinement input files and facilitate refinement in a series of pre-defined steps in order to carry out a basic structural refinement.
The interface between DASH and the refinement packages will only become available once the relevant sections of the Configuration Window (Choose Options-> Configuration from the main toolbar) in DASH have been completed (see Configuration).
There are two ways to start the Rietveld refinement:
-
Using the solution from a simulated annealing run.
-
With the crystal structure from e.g. a .cif or a .res file.
The first option is recommended. Note that it is possible to save all solutions from the simulated annealing and to read them back in at some other time (see Files saved from multiple runs of SA).

In order to do a Rietveld refinement for a crystal structure that is not a simulated annealing structure from DASH, choose Rietveld refinement in the main Wizard window. The following window will appear:

It is also possible at this point to start an SA structure solution run using the given crystal structure coordinates, without randomisation of initial coordinates, by clicking on the Start SA button.
Note that there can be ambiguities with respect to unit-cell settings and space-group settings: DASH only knows one setting for P212121, and loading a crystal structure file with a different space group setting will produce erroneous results.
The Rigid-Body Rietveld Refinement dialogue box can be accessed from the Analyse Solutions dialogue box after a simulated annealing run:

Refinable parameters are:
-
Positions and orientations of all Z-matrices.
-
Torsion angles.
-
Valence angles.
-
Bond lengths.
-
Global isotropic temperature pre-factor.
-
Preferred orientation (if used during simulated annealing).
In order to refine a variable, its check box must be selected. The check boxes for positions/orientations, torsion angles, valence angles and bond lengths are labelled V for variable and can be switched on and off either individually or as a group by using the Clear and Set buttons.
Due to the low x-ray scattering power of Hydrogen, torsion angles, valence angles and bond lengths involving one or more Hydrogen atoms are hidden by default and not refined.
The global isotropic temperature pre-factor is a factor that pre-multiplies the isotropic temperature factors of the individual atoms. This allows different atoms (e.g., different elements) to have different isotropic temperature factors.
Buttons
-
Calculate: calculates the powder pattern with the current parameters without refinement.
-
Save as... allows you to save the current crystal structure to a .pdb, .res, .cssr, .ccl or .cif file, and allows you to save the current powder pattern (both experimental + calculated) to a .pro file.
-
Compare: displays the original crystal structure and the Rietveld refined crystal structure superimposed.
-
Refine: performs a Rietveld refinement.
-
Close: closes the window.
-
View: displays the Rietveld refined crystal structure.
-
Axis... allows you to specify an axis to include the March-Dollase preferred orientation model.
-
Relabel: pressing Relabel relabels all atoms in all Z-matrices. This is useful for identifying e.g. torsion angles if a molecular model used for building a Z-matrix did not have unique atom labels.
Note: It is possible to manually enter values for all refinable variables. These values will be effective immediately.
-
When the Rigid-Body Rietveld Refinement dialogue box is launched, the Global isotropic temperature factor check box is automatically selected, and hitting Refine will carry out the first step of the Rietveld refinement. This step will not change the crystal structure, only the thermal parameters are affected.
-
The two most likely remaining candidates for refinement are the valence angles and the torsion angles. The temperature factors tend to correlate with all other parameters, and it therefore best not to refine the temperature factors in combination with other parameters. Therefore, the next step is to deselect the Global isotropic temperature factor check box and to click on the Set button for the valence angles, which will select all angles (except those involving Hydrogens) to be refined.
-
Next, deselect all angles by pressing the Clear button, then select all torsion angles (Set) and refine those.
-
Depending on how much the Chi2 values have changed, it may now be necessary to refine the positions and orientations of the Z-matrices again (keeping all bond lengths, angles and torsion angles fixed).
-
Depending on the quality of your data (it must be good) and the quality of your initial model (if you have reason to suspect it contains errors), you may try to refine some or all of the bond lengths.
-
Depending on how much the Chi2 values changed, you can return to the isotropic temperature factor again, and repeat the whole cycle.
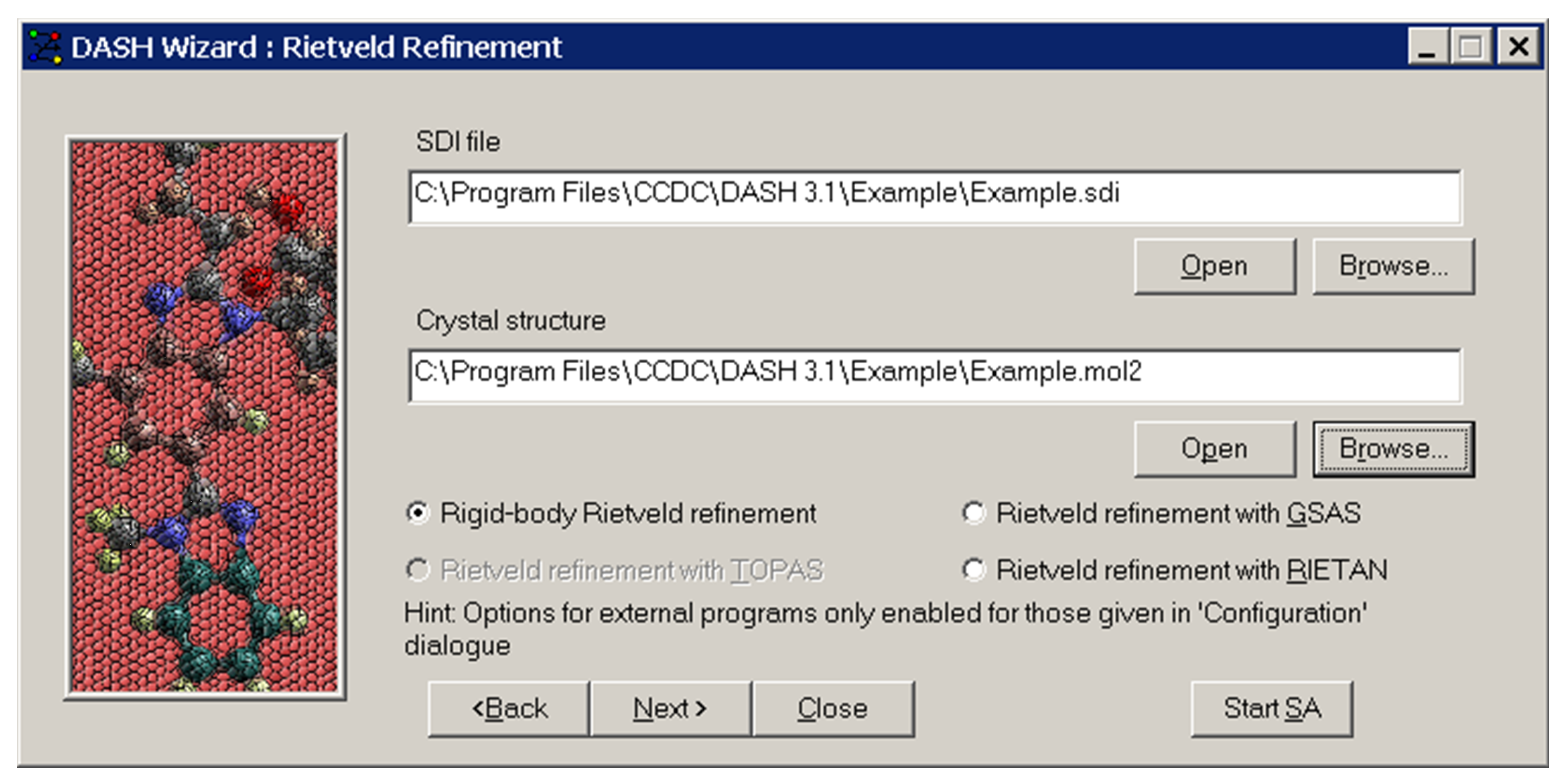
After choosing the refinement package you wish to use within the Rietveld Refinement Wizard and selecting the required .sdi and crystal structure files, click Next >.

In order to perform the Rietveld refinement, it is necessary to do a Pawley or Le Bail fit within your chosen refinement package using an input file generated by DASH. It may be advantageous to use a different data set or data range for the Rietveld refinement to that used for structure solution, so it will now be necessary to read in the diffraction data, click Next >.

Click Browse to find the diffraction data file that you want to use for Rietveld refinement and then click on Next > to read the data into DASH.

-
Check that the radiation type and wavelength specified are correct.
-
If the data are monochromated, ensure that the Monochromated check box is ticked.
-
If you are going to use GSAS for the refinement you have the opportunity to load in a GSAS .ins file. You may want to do this if you have measured instrument parameters that you would like to use in the refinement. Check the Load .ins file check box and browse to the location of the file.
-
Click Next > to continue.

The data range to be used can be modified at this point, but it is usual to utilise as much of the diffraction pattern as possible during Rietveld refinement, hence the data truncation option has been switched off by default. If you wish to truncate the data, enter the range to be used.
Click Next > to proceed.

Complete the steps of data preparation given (see Preparation of data for Rietveld Refinement using TOPAS, GSAS or RIETAN). The final step before sending the pattern to TOPAS is the Background Subtraction. This can be done within DASH, but it is usually better to allow TOPAS to perform the background subtraction (using 20 background terms as default).
Click Next >. A dialog will inform you when the first .inp file has been written and hence is ready to be loaded into TOPAS.

Once TOPAS is open, launch the Kernal Mode (Launch-> Kernal Mode) and set the .inp file to the .inp file written by DASH (Launch->Set INP file). To run the refinement, hit the <play> button (circled in red in the following screenshot) in the window called Launch Mode.

Once the cycle is complete, TOPAS will ask you whether you wish to update the .inp file with an .out file. Hit No and return to DASH.
The DASH interface for TOPAS will now guide you through the process of Rietveld refinement. At any point you may exit the refinement process by clicking on Close.

The Rietveld Refinement with TOPAS wizard window will be displayed with the Anisotropic broadening check box ticked. This is the first parameter to be refined.
-
Hit Write and a new .inp file will be written, ready to be loaded into TOPAS.
-
Return to TOPAS and hit the <play> button of the Launch Mode window. You will be asked whether you wish to update the .inp file with the .out file. Hit No and return to DASH.
-
Repeat the above cycle until all the parameters have been refined (six .inp files should be written for a standard refinement). In the final cycle a .cif file of the solution will be written by TOPAS.
Note: Default values for Weights on Restraints are provided but can be customised.

Complete the steps of data preparation given (see Preparation of data for Rietveld Refinement using TOPAS, GSAS or RIETAN). The final step before sending the pattern to GSAS is the Background Subtraction. This can be done within DASH, but it is usually better to allow GSAS to perform the background subtraction (using eight background terms as default).
-
Click Next > to save the GSAS .exp file. The Pawley fit will now automatically be performed within GSAS, follow the on-screen instructions when prompted. Once the fit has been done, a plot of the diffraction profile, the fit and the difference profile will be shown.
-
Close this plot by clicking on File-> Quit. Return to DASH where you can continue with the GSAS Rietveld Refinement.

The DASH interface for GSAS will now guide you through the process of Rietveld refinement. At any point you may:
-
View the structure (e.g. in Mercury) by clicking on View.
-
Switch to using EXPGUI for refinement by clicking on Launch an EXPGUI.
-
Exit the refinement process by clicking on Close.
The right-hand side of the dialogue box shows the current refinement options - to start with only two boxes are ticked to show that only the Scale and Background terms will be refined.
-
Click on the Refine button and the .exp file will be automatically updated then submitted to GSAS for the first step of refinement. When the cycle of refinement has completed you will be returned to the DASH interface.
-
The Refine options have been updated such that a Uiso parameter will be refined in the next cycle of refinement. Continue this process of refinement until all the checkboxes have been ticked.
-
The final cycle of refinement will be followed by output of the final structure in .cif format.
Note: Default values for Weights on Restraints (FACTR) are provided but can be altered.

Complete the steps of data preparation given (see Preparation of data for Rietveld Refinement using TOPAS, GSAS or RIETAN). The final step, before sending the pattern to RIETAN, is to choose the number of terms to be used by RIETAN for the background subtraction; the default value is eight.
Click Next > to save RIETAN's .ins file. The pattern fitting will now be performed automatically within RIETAN. Once the initial fit has been done, a plot of the diffraction profile, the fit and the difference profile will be shown in gnuplot graph. The fitting of the pattern is a two-step process: closing the gnuplot window (File->Exit) will start the second step of the fitting. Once complete, RIETAN will prompt you to save the .ins file.

The DASH interface for RIETAN will now guide you through the process of Rietveld refinement. At any point you may:
-
View the structure (e.g. in Mercury) by clicking on View.
-
Exit the refinement process by clicking on Close.
The right-hand side of the dialogue box shows the current refinement options - to start with, three boxes are ticked, showing that only the Scale, Background and Biso terms will be refined.
-
Click on the Refine button and the .ins file will be automatically updated, then submitted to RIETAN for the first step of refinement. When the cycle of refinement has completed you will be prompted to save the .ins file before being returned to the DASH interface.
-
The Refine options will have been updated to include the atomic coordinates for the final cycle of refinement. Click Refine to complete the refinement.
Note: Default values for the Number of cycles, Penalty parameter (TK) and TK increasing multiplier (FINC) are provided but can be altered.
Note: Output from RIETAN is written to a .lst file should you wish to look up the reported Rwp and Rp values for the refinement cycles.